
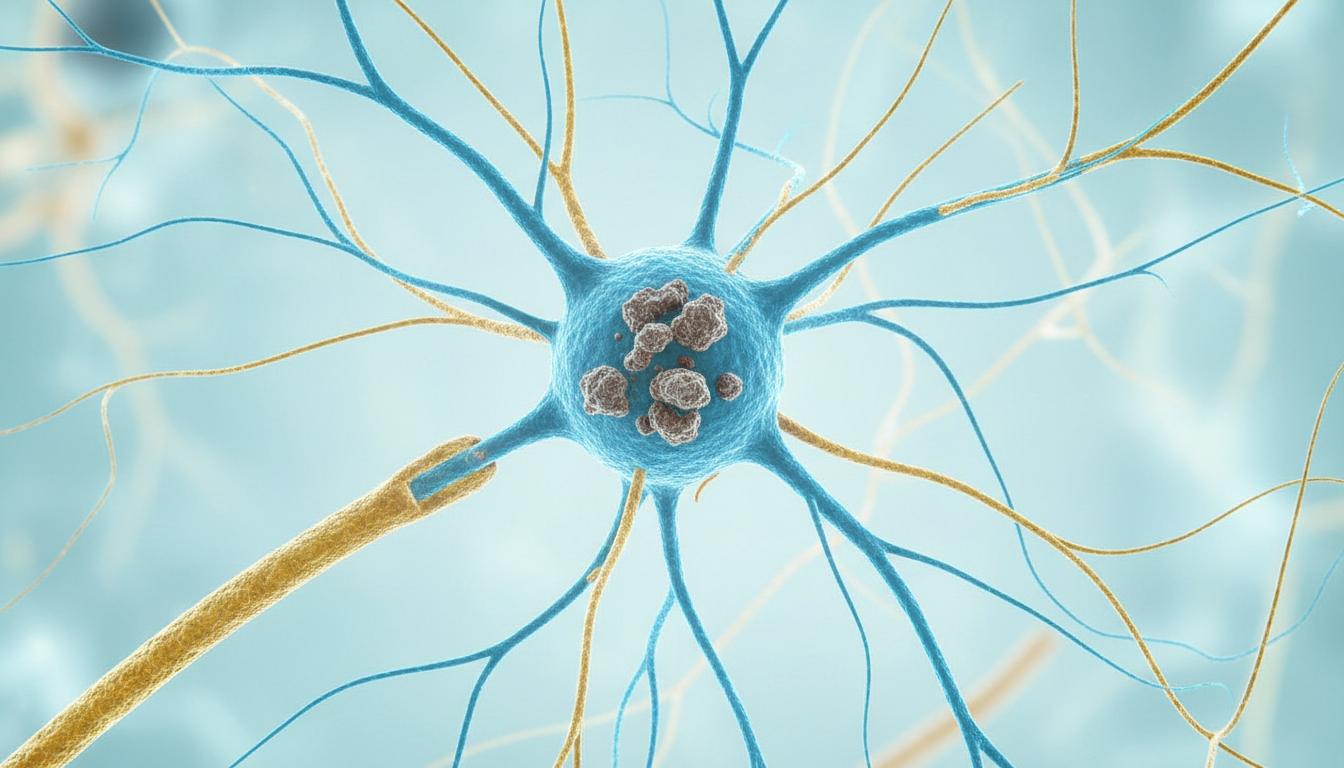
Have you ever heard of a condition called GM1 gangliosidosis? Probably not. Because it's a rare condition, it doesn't affect everyone. But it's important to be aware of these conditions. Simply put, this disease causes certain particles in our bodies, especially nerve cells, to accumulate and damage the brain and spinal cord. This is irreversible damage.
What is GM1 gangliosidosis?
Okay, let's go into a little more detail. GM1 gangliosidosis is a rare genetic disease . It causes certain molecules in our bodies, especially fats and sugars, to build up inside nerve cells in the brain and spinal cord. This build-up occurs because the body doesn't produce a special enzyme that helps break down these molecules. When these molecules build up, the nerve cells become damaged and lose their function.
This disease is inherited . This means that it is caused by a mutation in the genes inherited from both parents. Symptoms may begin as a baby, or may appear in childhood, or even later in life. This is a disease that belongs to a group called lysosomal storage disorders . Unfortunately, there is currently no cure for this disease.
What are lysosomal storage disorders?
Now you're probably wondering, "What is this lysosomal storage disease?" Let's explain that too.
Lysosomal storage disorders are a group of inherited diseases that affect our metabolism. You know, our metabolism is the process by which we convert the food we eat into energy and remove toxins from the body. There are about 50 types of lysosomal storage disorders. For example, Tay-Sachs disease is one such disease.
"Lysosomal" refers to the small compartments inside our cells, which are called lysosomes. Inside these lysosomes are special proteins called enzymes. These enzymes break down large molecules like fats and sugars that enter our bodies and turn them into simpler molecules. However, in the body of a person with a lysosomal storage disease, these enzymes cannot do that job properly. Then those large molecules do not break down and accumulate inside the cells. That is why it is called a "storage disorder."
Lysosomal storage diseases like GM1 gangliosidosis are progressive diseases . That is, as the amount of these molecules accumulate in the body, the symptoms gradually become worse.
What are the main types of GM1 gangliosidosis?
GM1 gangliosidosis is a congenital disease. This means that the genetic change that causes the disease is present at birth. However, it can take some time for symptoms to appear. Doctors classify the disease according to the age at which symptoms first appear. Sometimes the symptoms and timing of these types can overlap.
There are three main types:
1. Classic infantile (Type 1): Symptoms usually begin to appear around 6 months of age. This type becomes severe very quickly.
2. Juvenile (Type 2 - Juvenile): In this type, symptoms usually appear between the ages of 1 and 5. The disease progresses more slowly than in the first type.
3. Adult (Type 3 - Adult): Symptoms can begin as young as 3 years old, or as late as 30 years old. The disease progresses more slowly than the other two types.
How common is this disease?
GM1 gangliosidosis is an extremely rare disease . Worldwide, the disease affects only a very small number of people, approximately 1 in 100,000 or 1 in 200,000 .
What causes GM1 gangliosidosis?
The main cause of this disease is a mutation in the GLB1 gene . This GLB1 gene helps to make an enzyme called beta-galactosidase, which is found in our lysosomes. This enzyme breaks down molecules like GM1 ganglioside. This GM1 ganglioside molecule is very important for the proper functioning of nerve cells in our brain.
Because of that genetic change, the body is unable to break down the GM1 ganglioside molecule. Then these molecules gradually start to accumulate in tissues and organs. This causes irreversible damage to the cells of the nervous system, especially the brain and spinal cord .
Who is at higher risk of developing this disease?
To develop GM1 gangliosidosis, a child must inherit the mutated GLB1 gene from both parents . In this case, both parents are carriers of the gene mutation, but they do not develop the disease. Doctors call this an autosomal recessive disorder .
Even if both parents are carriers of the GLB1 gene mutation, their children may or may not develop the disease. If they do, the child will usually develop the same type of disease that was inherited by previous generations.
If both parents have this gene mutation, each of their children has the following chances:
- There is a 1 in 4 chance of not having the mutated gene and not being at risk of disease.
- GM1 gangliosidosis has a 1 in 4 chance of developing the disease.
- There is a 1 in 2 chance of not developing the disease, but of being a gene carrier.
Although this genetic mutation can run in any family, Japanese people are more likely to develop type 3 diabetes .
What are the symptoms of GM1 gangliosidosis?
The symptoms of GM1 gangliosidosis vary depending on the type. Also, some symptoms may be common to several types.
Characteristics of Classic Infantile (Type 1):
- Distended abdomen
- Enlarged spleen and enlarged liver
- Extreme startle response to loud noises
- Hearing loss
- Red spots on the eyes and loss of vision
- Regression of developmental milestones - For example, a baby who was once able to smile or hold their head up can no longer do those things.
- Seizures
- Stiff joints or skeletal abnormalities
- Weak muscle tone (hypotonia)
Characteristics of Juvenile (Type 2):
- Ataxia - coordination and balance problems
- Corneal disease - clouding
- Difficulty swallowing (dysphagia)
- Dystonia - excessive muscle contractions
- Loss of cognitive function or thinking skills
- Problems with speech (dysarthria)
- Seizures
Characteristics of adults (Type 3):
- Muscle weakness or atrophy
- Corneal disease - clouding
- Muscle spasms (Dystonia)
- Noncancerous skin lesions
How is GM1 gangliosidosis diagnosed?
If someone in your family has the disease, prenatal testing can help determine whether your unborn baby has the gene mutation. This can be done through a genetic amniocentesis or chorionic villus sampling (CVS) test. These can detect cells that contain the mutation.
In addition, these tests are performed to diagnose this disease in children from infants to adults:
- Enzyme assay: This measures the amount of beta-galactosidase enzyme in your blood.
- Molecular genetic test: This is also a blood test. It checks DNA sequences to identify the GLB1 gene mutation. As you know, DNA (deoxyribonucleic acid) is what we inherit from our parents.
- Newborn screenings: In some countries, routine newborn screening in hospitals includes enzyme tests for lysosomal storage disorders.
What are the treatments for GM1 gangliosidosis?
There is currently no specific treatment, surgery, or cure for GM1 gangliosidosis. Treatment is focused on managing the individual's symptoms and maintaining a good quality of life . For example, a person with seizures may be given a ketogenic diet (keto diet) or anticonvulsant drugs such as gabapentin to control their seizures.
However, medical researchers are continuing to find new ways to treat and even prevent the disease. You or your child may also have the opportunity to participate in clinical trials that test new treatments that are still in the research phase.
These experimental treatments may include:
- Enzyme enhancement or enzyme replacement therapy
- Gene therapy
- Stem cell transplants (also called bone marrow transplants)
- Substrate reduction therapy - This attempts to stop the disease process by changing the molecules that are being synthesized.
Can GM1 gangliosidosis be prevented?
If you are a carrier of the mutated gene that causes GM1 gangliosidosis, you can talk to a genetic counselor to discuss options that can reduce the chance that your children will inherit the gene.
For example, a procedure called Preimplantation Genetic Diagnosis (PGD) can identify embryos that do not have the mutated gene. The doctor can then transfer those healthy embryos into the uterus using a procedure called In Vitro Fertilization (IVF) . PGD can help ensure that your child is not a carrier of the gene or will not have the disease.
What will the future life be like for someone with this disease?
The symptoms of GM1 gangliosidosis gradually worsen over time. The life expectancy and quality of life of a person with this disease vary depending on the type of disease:
- Babies with the first type (Type 1 - classic infantile) can probably live for about 2 years .
- Children with Type 2 (juvenile) may live into mid-childhood or early adulthood , depending on the age at which symptoms begin.
- People with Type 3 (adult) have a shorter lifespan. This varies depending on the age at which symptoms begin, the nature and severity of the symptoms.
When should you see a doctor?
If you or your child has any of these symptoms, see your doctor immediately:
- Balance or gait problems
- Difficulty breathing, swallowing or speaking
- Hearing or vision changes
- Red spots on the eyes
- Seizures
What should you ask your doctor?
You might want to ask your doctor questions like these:
- What type of GM1 gangliosidosis do I (or my child) have?
- What medications are available to relieve symptoms?
- What can we do to relieve symptoms at home?
- What kind of medical specialists should we see?
- Should I watch out for signs of complications?
- Should other members of my family be tested for this genetic mutation?
Finally, take-home message
GM1 gangliosidosis is a rare, inherited disease that causes the body to be unable to break down fat and sugar molecules. It belongs to a group of lysosomal storage disorders. When these molecules build up, symptoms such as seizures, balance problems, and difficulty swallowing occur.
To develop the disease, you must inherit the gene mutation that causes the disease from both your mother and father. Treatments are aimed at relieving specific symptoms. Although there is currently no cure, clinical trials are underway for new treatments. You can talk to your doctor about ways to reduce the risk of passing on this gene mutation to future generations.
It's normal to feel scared and anxious when you learn about a condition like this. However, it's important to get the right medical advice and support . You are not alone, and there are doctors and loved ones to help you on this journey.
` GM1 gangliosidosis, genetic diseases, lysosomal storage diseases, neurological diseases, rare diseases, beta-galactosidase, GLB1 gene
💬 අදහස් (0)
තවමත් කිසිදු අදහසක් පළ කර නොමැත. ඔබේ අදහස පළමු වරට මෙහි එක් කරන්න.
ඔබේ අදහස එක් කරන්න